Description #
This module is designed to help students and clinically registered nurses develop a better understanding of the skills involved in pre code management.
Learning Objectives #
Upon completion of this module the learner should be able to:
1. Understand the etiology and pathophysiology of Acute Coronary Syndrome (ACS), heart failure (HF) & cardiogenic shock
2. Identify early indicators of impending or developing ACS, HF & cardiogenic shock
3. Relate diagnostic indicators to clinical indicators for ACS, HF & cardiogenic shock
4. Initiate and implement appropriate early interventions for ACS, HF & cardiogenic shock to minimize further deterioration
5. Establish a systematic approach to report these cardiac events to appropriate health care team
6. Implement appropriate emergency responses and contribute to the resuscitation of the cardiac patient as a member of the health care team
Part I: Acute Coronary Syndrome (ACS) #
1. Etiology and Pathophysiology of Acute Coronary Syndrome (ACS)
2. Early indicators of impending or developing ACS
3. Relate diagnostic indicators to clinical indicators of ACS
1. Etiology and Pathophysiology of Acute Coronary Syndrome (ACS) #
Atherosclerotic plaque build-up occurs when the endothelial layer of the coronary artery wall becomes injured. Excess fat and cholesterol in the blood gets deposited in the artery wall. Plaque which has a lipid rich core and a thin fibrous cap forms as the fat deposits build up. As the plaque size increases, blood supply to the myocardium decreases resulting in cell injury and death. Plaque ruptures may occur due to mechanical injury, inflammation and infection. When an arterial plaque ruptures, vasospasms may occur thus further narrowing the coronary artery. See Figure 1.

Formation of atherosclerotic plaque is associated with certain risk factors. The risk factors are divided into modifiable and non-modifiable (Table 1).
Table 1. Risk Factors for Atherosclerotic Plaque formation.
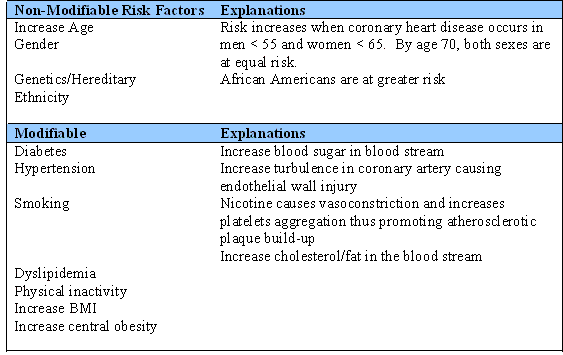
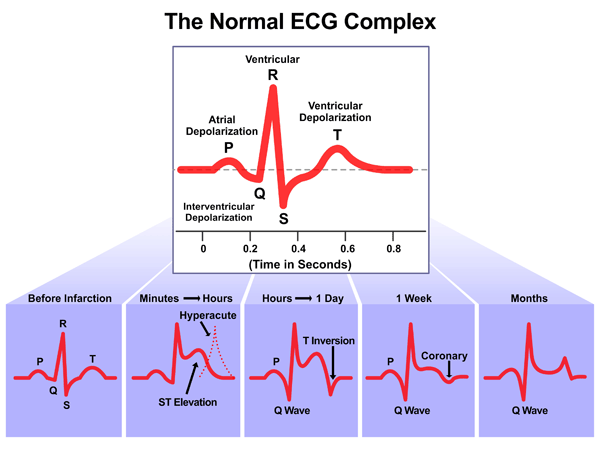
ACS consists of a spectrum of heart conditions that cause myocardial infarction (MI). These conditions include unstable angina (U/A), Non ST-Elevation MI (NSTEMI) and ST-Elevation MI (STEMI). In U/A, a thrombus may be partially blocking the artery intermittently, thus, causing chest pain/discomfort at rest and/or activity. On a 12-lead electrocardiogram (ECG) with ischemia, ST depression and T wave inversion are often seen (See Figure 2). A normal T wave is usually in the same direction as the QRS except in the right precordial leads.
Total occlusion of the coronary artery results in ST elevation on the ECG. Total occlusion leads to myocardial cell death which can be detected by the release of the biochemical markers, troponin and CK-MB, into the circulation. The ultimate goal of treatment is to prevent permanent cell death; thus, reperfusion is the primary intervention for selected patients with acute MI whenever possible.
Figure 2 Stages of ST changes in ACS
2. Early indicators of impending or developing of ACS #
- Classic symptoms of ACS present as retrosternal chest pain that may or may not radiate to the jaw, neck, arms and/or back. Usually the patient describes the chest pain as a discomfort, heaviness, pressure, aching, crushing, burning or squeezing. This pain may be intermittent but usually lasting > 15 minutes (Devon & Ryan, 2005). There are other associated symptoms with the pain and these may include dyspnea (shortness of breath), diaphoresis, syncope, epigastric pain and nausea.
- Women with ACS are often misdiagnosed because they do not always complain of chest pain. Only 29% of women report having chest discomfort (McSeeney et al, 2003). The typical symptoms women would report are weakness, fatigue, sleeps disturbance and dyspnea.
Every patient may present with different symptoms of chest pain. Showing the hallmark symptoms of chest pain will expedite treatment. Those who do not show the typical symptoms often fail to recognize the seriousness of their symptoms, thus delay reporting or seeking treatment and/or being misdiagnosed.
3. Relate diagnostic indicators to clinical indicators for ACS #
Clinical assessment of patients should include:
- A clinical history
- ECG findings
- Cardiac markers
- A physcial examination
- A chest X-ray
Early assessment of a patient with chest pain is critical, especially in the first few hours after the onset of symptoms. An ECG can determine whether the patient has a STEMI or a NSTEMI. The former diagnosis will benefit most from reperfusion (by thrombolysis or coronary angioplasty). If angioplasty is not immediately available, thrombolytic therapy should be initiated within 30 minutes unless the risk of treatment is greater than potential benefits.
Patients with NSTEMI will benefit the most with cardiac monitoring and administration of ASA and an antithrombin agent (heparin, LMW heparin) and antiplatelet drugs (glycoprotein IIb/IIIa inhibitor) and/or possible early revascularization (angioplasty).
Biochemical Markers #
Cardiac markers are a vital part of diagnosing acute myocardial infarction. Creatine kinase (CK), myoglobin (MB) and CK-MB were initially used to diagnose patients with ACS until troponin was discovered. CK is found in muscles and tissues of the brain, kidney, lung, and gastrointestinal tract. MB is found in cardiac and skeletal muscles. As a result, CK and MB levels could be affected by non cardiac conditions such as trauma, seizures and renal insufficiency. CK-MB is more cardiac specific and is often used in conjunction with troponin.
Troponin I and T are the preferred choice because they are more selective in finding myocardial injury for it is specific to the myocardium. Troponin is so sensitive that it is not detectable in plasma of healthy patients. Positive cardiac troponin level is an indicator for recurrent thrombotic episode and suggests the need for aggressive antithrombotic management. Levels are normally drawn at the time of symptom onset and repeated 6 to 8 hours after. Troponin levels may remain up for two weeks.
Patients with positive cardiac markers “have a 5 fold higher risk of early and later reinfarction and death than possible ACS patients without biochemical evidence of myocardial injury” (Fitchett, Goodman, & Langer, 2001, p. 1311).
Treatment of ACS #
The initial assessment of chest pain is only the start of a treatment cascade for ACS. Treatment of ACS depends upon the results of the 12 Lead ECG, and whether the changes on the ECG show ST depression, elevation or no change at all. Initial treatment will include morphine, oxygen, nitroglycerin & ASA (acronym: MONA). Other treatments for ACS may include administering Heparin IV, Beta Blockers, and glycoprotein (IIb/IIIA) inhibitors. Be prepared to order further labwork (cardiac markers, CBC, electrolytes) and organize further diagnostic testing.
The patient will likely need a diagnostic cardiac catheterization, and then may require percutaneous cardiac intervention (PCI) or cardiac surgery.
Part II: Heart Failure (HF) #
HF is on the rise, as the Canadian population ages and survival rates for ACS or MI increase. Because it is a progressive condition and is often not reversible, the incidence of HF is increasing among hospitalized patients. Therefore, it is important to identify patients at risk of developing HF, and to intervene before it progresses to refractory heart failure or cardiogenic shock. Heart failure (HF) is an acute or chronic condition that occurs when the heart is unable to effectively pump blood. Acute HF is the sudden onset of symptoms over hours to days that usually require emergency intervention. If the acute symptoms are no longer reversible, the heart failure becomes chronic. Chronic HF is the gradual development of symptoms over months to years.
Etiology and Pathophysiology of heart failure #
Heart failure is a complex syndrome that develops from both cardiac and non-cardiac diseases. The primary cardiac causes of HF include diseases of the pericardium, myocardium, valves or great vessels. Acute coronary syndrome (ACS) is the most common cause of HF, as it affects the contractile function of the myocardium secondary to ischemia or necrosis. Secondary causes of HF include conditions such as unrecognized volume overload, excess dietary salt or fluid intake. Non-cardiac diseases that may cause HF include conditions that require increased cardiac output (CO) or increased resistance to output. Regardless of the etiology, the CO is insufficient to meet the metabolic demands of the body at rest or/and on exertion.
Table 2 Examples of conditions leading to heart failure

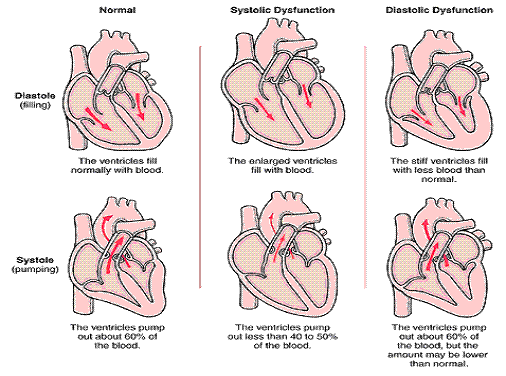
Traditionally, HF has been classified as low or high cardiac output failure based upon the heart’s ability to pump an adequate amount of blood during a cardiac cycle. Unfortunately this simple classification is inadequate, and cannot describe the true complexity of the clinical syndrome. Therefore, HF is now further differentiated into impaired contractility (systolic), impaired filling (diastolic) or both dysfunctions to provide a better understanding of the pathophysiology of a failing heart. Here are some common terms used in relation to heart failure:
a. Low output failure constitutes 90% of all documented cases of HF. The weakened heart is unable to pump enough oxygenated blood to perfuse the body’s tissues adequately, and thus cannot meet the body’s metabolic needs.
b. High output failure indicates that cardiac function is normal, or even high, but the metabolic demands of the body are so high that the heart cannot meet that need. Sepsis and hyperthyroidism are the most common causes of high output failure.
c. Left ventricular (LV) dysfunction is a very common cause of heart failure. Because the left ventricle pumps blood to the systemic circulation, any dysfunction that occurs (whether because of myocardial infarction or cardiomyopathy, or by LV dysfunction) will cause low cardiac output as well as pulmonary congestion.
d. Right ventricular (RV) dysfunction occurs if the right ventricle is damaged by MI or by pulmonary hypertension (the right ventricle cannot pump against very high pressures like the left ventricle). RV failure will cause a backup in systemic circulation that is often evidenced by peripheral edema.
e. Biventricular dysfunction generally occurs when LV dysfunction is not treated in a timely fashion, and metabolic needs of the body are not met. The patient will display symptoms of both RV and LV dysfunction, including peripheral edema and pulmonary congestion.
f. Systolic dysfunction refers to an impairment in the LV’s ability to contract effectively during systole. The leading cause of systolic dysfunction is ACS, since it causes decreased blood supply to the myocardium. Chronic hypertension and aortic valve stenosis also increase the resistance the LV must overcome in order to eject blood effectively.
g. Diastolic dysfunction occurs when the left ventricle becomes stiff and unable to relax effectively during diastole. 40-50% of people with HF display some degree of diastolic dysfunction. The major cause of this is advanced age, as the ventricle stiffens and becomes less compliant with age. Other causes include Valvular heart disease, untreated hypertension, and coronary ischemia.
Figure 3 Types of Heart Failure
Compensatory Mechanisms of Heart Failure #
In the early stages of heart failure, the body attempts to compensate for the failing pumping action of the heart. The cardiac response initially stretches the left ventricle, to accommodate the increased volume left behind in the left ventricle (LV) after systole. The LV also becomes thicker, which ultimately increases myocardial oxygen demand.
The clinical manifestation of low cardiac output is hypotension. The body senses this change and attempts to increase the systemic blood pressure by triggering the sympathetic nervous system, neurohormonal compensatory mechanisms, and the renin-angiotensin-aldosterone system. All of these systems serve to temporarily increase the blood pressure by retaining fluid and causing vasoconstriction. Initially these mechanisms will be successful, but in the long run are more damaging, as they further increase the metabolic demands upon the failing heart, with an increase in cardiac workload, preload and afterload.
The progression of HF is a result of the interaction between the compensatory mechanisms of the brain, heart, kidneys and periphery. As the body continues to attempt to compensate for the failing ventricles, cardiac output continues to drop, and the LV stretches to accommodate the increased volume, and stiffens to pump more forcefully.
Fluid begins to build up in the lungs as it backs up from the LV. The RV also becomes affected as the build up of fluid in the lungs backs even further. This causes a chain reaction of congestion throughout the body, and results in low arterial oxygen values. Long term untreated heart failure can also cause liver congestion and ascites. Dyspnea is a common finding in heart failure, as the body attempts to compensate for the low oxygen levels in the blood.
Progressing to Biventricular Heart Failure #
In LV systolic dysfunction a the CO decreases due to poor contractility, as the LV cannot empty its blood volume; it starts to dilate to accommodate the larger blood volume.
In contrast, LV diastolic dysfunction a the LV is too stiff to allow adequate filling during diastole, to maintain CO, the LV pressure increases resulting in LV hypertrophy. As the LV enlarges, the left atrium is unable to empty its blood volume into the LV, resulting in blood backing into the pulmonary venous system causing an elevation in the pulmonary pressure.
Normally pulmonary pressure is a low-pressure system but when the pulmonary capillary (hydrostatic) pressure exceeds the plasma protein (oncotic) pressure, fluid shifts from the capillaries into the interstitial space and alveoli of the lungs. This changes the compliance of the lungs causing increase work of breathing. Lymphatic drainage in the lungs cannot compensate for the increase in pulmonary fluid leading to pulmonary edema and changes in ventilation/perfusion relationships. As the deoxygenated blood from the pulmonary artery capillaries pass through the fluid filled alveoli it cannot pick up any oxygen resulting in low arterial oxygen (PaO2). Dyspnea is a common finding in HF, because low arterial oxygen stimulates the respiratory centre to increase the respiratory rate to get more oxygen into the body, resulting in respiratory alkalosis. The hypoxic vasoconstriction and elevated pulmonary pressure result in increased afterload for the RV. The right ventricle, a smaller ventricle with less muscle mass, is unable to empty its blood volume when there is resistant to outflow; thus, the pressure in the RV starts to increase.
When the RV pressure is greater than the right atrium pressure, the systemic venous circulation will increase. As a result of blood backing into the systemic venous system, fluid will shift into tissue such as in the ankles, feet, and abdominal viscera. Heptomegaly is a common finding in RV failure, as the congestion in the liver worsens, it affects liver function resulting in elevation of liver enzymes, ascites and impaired metabolism of aldosterone, which further promotes fluid accumulation. Prolong venous congestion in the viscera can cause GI symptoms such as anorexia, malabsorption, and enteropathy, resulting in muscle wasting, anemia and fatigue.
Low BP triggers the activation of SNS and RAAS to maintain perfusion to vital organs. The increase in preload and afterload from sodium and fluid retention and vasoconstriction increase the circulating volume and systematic vascular resistant for the LV. These changes increased workload of the heart and will result in increase myocardial oxygen consumption and might result in myocardial ischemia with further deterioration of CO. When persistent hypoperfusion to vital organs occurs, there is a decrease in oxygen delivery and the body revert into anerobic metabolism. Lactic acid is the by-product of anaerobic metabolism and accumulation of this acid will result in lactic acidosis. Acidosis will result in muscle fatigue and diminish response to catecholamine. The end result is further decrease in CO and ongoing activation of the compensatory mechanisms. If this process of forward and backward failure persists, the patient will go into refractory heart failure and cardiogenic shock.
2. Early indicators of impending or developing HF #
HF affects all body systems by the time clinical signs and symptoms occur and the ventricular remodelling has already occurred with cardiac dilatation, hypertrophy and spherical rotation of the heart. The early signs and symptoms of HF are subtle and may be non-specific.
Figure 1: Common Signs & Symptoms of HF
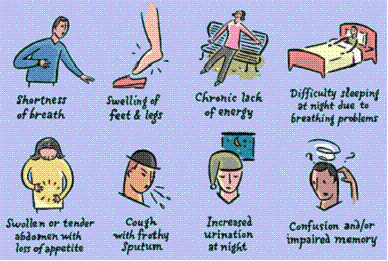
Signs and Symptoms of Heart Failure Early Late Mechanism

3. Diagnostic & clinical indicators for HF #
In 2001 American Heart Association (AHA) and American College of Cardiology (ACC) identified four stages in the development of heart failure. The purpose of the staging system is to identify high-risk patient of developing HF and preventing LV remodelling by early treatment. Patient progresses through these stages in one direction and does not revert back to earlier stages.

Another classification system that might help to determine the severity of HF is the New York Heart Association (NYHA) functional classification system. The focus is on dyspnea. Dyspnea is a common symptom of HF and the level of exertion required to cause symptoms reflects the degree of cardiac function on how well the HF is managed. However, the correlation between cardiac function and symptoms is poor, as dyspnea is a subjective evaluation.

In diagnosing HF, taking a complete patient’s history is vital, as this will identify the risk factors, the functional ability and the cause of HF. The clinical assessment and diagnostic tests will confirm the presence of heart failure and establish the severity of HF based AHA/ACC and NYHA.
Clinical assessment should include checking for (pitting) edema/ascites, jugular distention (indicative of right filling pressures), activity tolerance, and systemic circulation (e.g. capillary refill, peripheral pulses). Table 3 shows the common diagnostic tests used to confirm HF.
Table 3 Diagnostic Tests Used to Diagnose Heart Failure
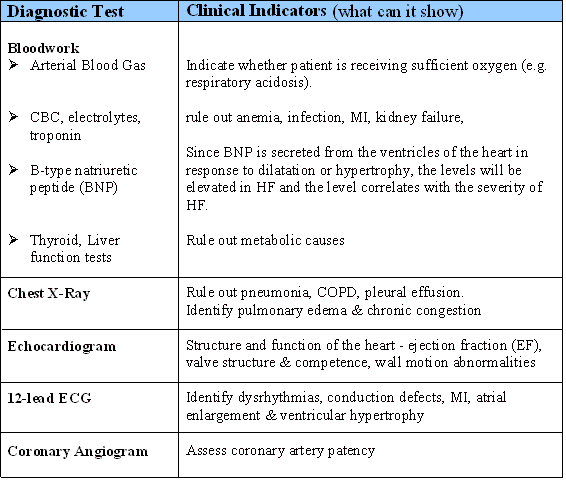
4. Initiate and implement appropriate early interventions for HF to minimize further deterioration #
The treatment goal for HF is to treat the underlying cause and symptom management. The immediate nursing interventions for these patients follow the principle of ABC for acute and chronic phase of HF. Prior to the physician arriving, nurses can immediately do the following interventions:

Airway (A)
Ensure the patient’s airway is patent
Breathing (B)
Position the patient in high fowler’s position (if patient is not hypotensive) Apply oxygen until the oxygen saturation is greater than 92% Call RT if the patient’s saturation does not improve with oxygen via facemask at 10L/min (May consider BIPAP to reduce pulmonary edema and respiratory effort) Monitor the oxygen saturation via pulse oximetery continuously Re-assess respiratory rate and pattern
Circulation (C)
Monitor the patient’s BP and HR Ensure the IV is patent Position patient with feet lower than the heart to decrease venous returns (if patient is not hypotensive)
Anticipate the following medications likely to be ordered:
A: ACE inhibitors or ARB to block the neurohormonal action & HTN
B: Beta-blocker (Metoprolol) for tachycardia and HTN (if patient does not have respiratory disease/asthma)
C: Calcium channel blocker for arrhythmias or intolerance to beta-blockers
D: Diuretics (Lasix) for pulmonary edema
N: Nitroglycerin patch for preload reduction and chest pain
- Continue to assess the ABC and stay with the patient to monitor for deteriorations
- Keep patient NPO
- Prepare for urinary catheterization if the patient does not have a catheter
- Keep the patient’s chart, VS flow sheet and lab results at the bedside
Part III: Cardiogenic Shock #
Cardiogenic shock is a condition of continued low C.O., despite adequate intravascular volume, that results in severely impaired tissue perfusion. Cardiogenic shock is occasionally referred to as pump failure, and can occur in nearly 5-10% of all patients who experience acute myocardial infarction (Hollenberg, Kavinsky and Parillo, 1999), with a mortality rate that in some populations can reach 50-80% (Bromet and Klein, as cited in Bench, 2004). In a study of patients experiencing acute MI, almost all of the patients who developed cardiogenic shock after their event did so within 48 hours of symptom onset (Ducas and Grech, 2003). In early stages, cardiogenic shock may resemble other forms of shock (e.g, hypovolemic shock), the compensatory mechanisms are hindered by pump failure, leading to a more rapid progression through the stages. It is imperative to identify the signs of cardiogenic shock early, because the earlier you identify the symptoms, the more likely it is that your patient will not experience irreversible organ damage.
Etiology and pathophysiology of Cardiogenic Shock #
Cardiogenic shock develops when the left ventricle has either directly or indirectly lost its ability to pump effectively. With the ventricle unable to deliver oxygenated blood to the rest of the body, tissues become hypoperfused and a vicious cycle of decompensation develops.
Any condition that decreases left ventricular function can cause cardiogenic shock, but the most common causes include:
- Acute MI
- Heart failure
- Cardiomyopathy
- Acute dysrhythmias
- Cardiac tamponade
- Valve dysfunction
- Papillary muscle rupture
- Massive pulmonary embolus
- Tension pneumothorax
As the left ventricle fails, a series of compensatory mechanisms are attempted in order to increase cardiac output and maintain organ function. Baroreceptors, located in both the aorta and carotid arteries, stimulate the SNS which in turn increases heart rate, left ventricular filling pressure and peripheral resistance (vasoconstriction). These actions all increase preload, or the venous return to the heart. Initially, the compensatory activation of SNS will stabilize the patient, but overtime, the failing heart will be unable to handle the increased venous return, increased workload of the failing ventricle(s) and increased oxygen demands. As a result, an ever increasing spiral of oxygen demand outpaces supply leading to further deterioration if action is not taken to reduce the cardiac workload. Specifically, as the oxygen supply dwindles, the body shifts from aerobic to anaerobic metabolism, causing an increase in lactic acid production. The liver then cannot adequately metabolize the lactic acid, further interfering with cardiac output.
See the figure below for a simple diagram that demonstrates the complex interaction among the physiological processes of cardiogenic shock:
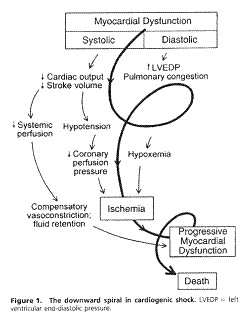
from Hollenberg, Kavinsky, & Parillo (1999). Cardiogenic shock, Annals of Internal Medicine,131(1) p. 49.
Early indicators of impending or developing cardiogenic shock #
One of the most important things to remember about cardiogenic shock is that early identification and treatment is vital. A good way of preventing the onset of cardiogenic shock is to watch closely for it in patients who have the physical conditions listed above. Your patient may have had a surgical procedure unrelated to his cardiac history, but the stress of the surgery may have been enough to cause decompensation.
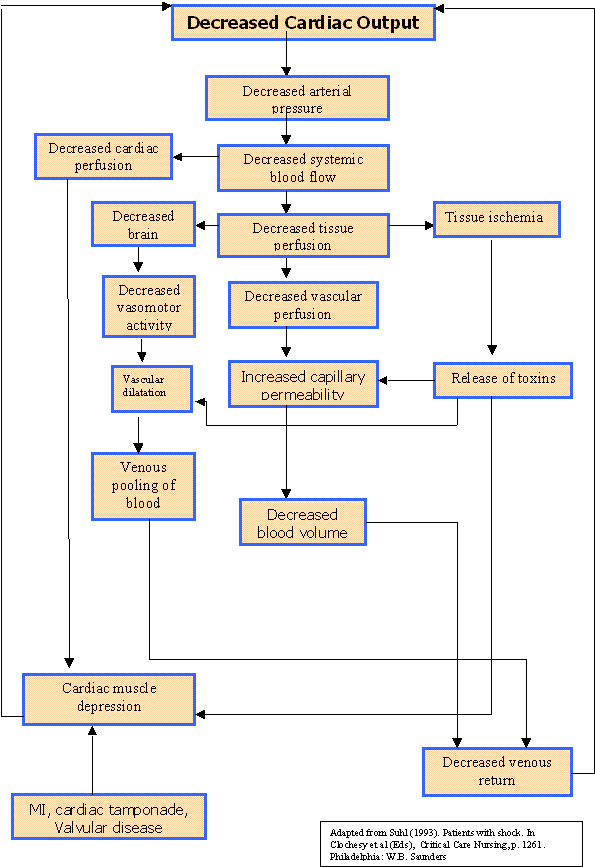
Cardiogenic shock is best defined by its three “cardinal signs”—low blood pressure, decreased level of consciousness, and decreased urine output. These signs all indicate that there is a general decrease in tissue perfusion, and the three body systems that show the earliest signs of inadequate oxygenation are the brain, heart and kidneys.
Diagnostic & Clinical Indicators for cardiogenic shock #
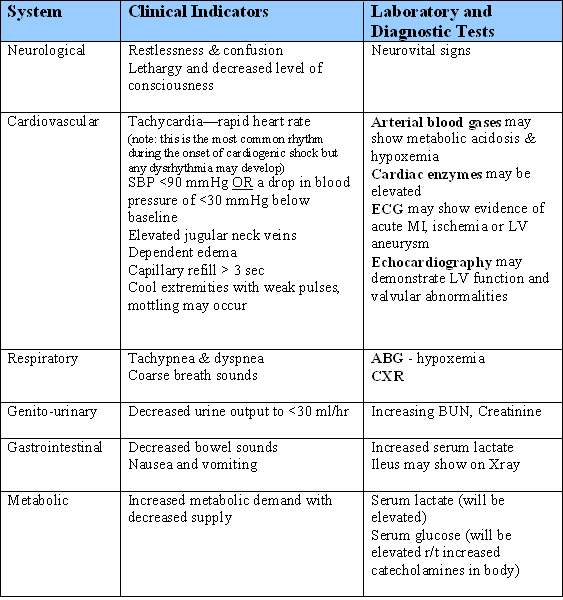
Often the patient will deteriorate very quickly from a situation of ACS or heart failure into shock, as the left ventricle fails and the body decompensates. However, here are the clinical signs that the patient is experiencing cardiogenic shock, as well as the laboratory findings that will assist with the diagnosis:
Early intervention for Cardiogenic shock: goals and treatment #
- The goals of treatment are to enhance cardiovascular status and hopefully reversing the shock state by increasing cardiac output, improving cardiac perfusion, and decreasing cardiac workload. This is often difficult to accomplish, because medications that treat one symptom of the shock state may cause complications that can further compromise the patient.
For example, a common medication to treat decreased ventricular contractility is dobutamine, which can increase the force of contraction of the left ventricle. However, dobutamine can worsen the patient’s tachycardia, which will eventually increase the workload on the heart.
- Treatment includes a combination of cardiovascular drugs and mechanical assist, both of which will be commenced in a critical care setting.
- Cardiogenic shock is a medical emergency, and a physician should be involved in the patient’s care as soon as possible. While it is often difficult to identify the shock process in the early stages, with its vague signs, it is imperative that early interventions be taken.
Remember, whenever you feel that you need a physician at the bedside immediately (i.e. the patient has deteriorated severely), call a Code Blue.

Using the ABC approach, here are some of the interventions you can take:
Airway (A)
- Ensure that the patient maintains a patent airway. This may mean that you have to insert an oral airway if they become obtunded or lose consciousness.
Breathing (B)
- Monitor oxygen saturations, respiratory rate and effort q15mins and prn
- Administer supplemental oxygen, probably by face mask, to achieve a SpO2 of >90%
- Call an RT for help with managing the patient’s oxygenation
- Monitor the patient’s breath sounds frequently for signs of increasing fluid overload as the left ventricle fails and fluid backs up into the lungs
Circulation (C)
- Monitor pulse and blood pressure q15mins, using a dynamap. Immediately report a drop in blood pressure to <90 mm Hg, or a drop of >30 mm Hg below baseline
- If a monitor is available on your unit, hook the patient up to the monitor and begin continuous cardiac monitoring.
- Place the emergency cart in the room
- Start at least two large bore IV lines for fluid and drug administration
- Administer diuretics as ordered by the physician—this will decrease preload on the heart and may increase cardiac output
Other:
- Insert a foley catheter and monitor urine output hourly
- Expect to draw serum electrolytes and a complete blood count. The RT may draw arterial blood gases to assess oxygenation, ventilation, potassium level and acid-base status
- Prepare the patient for transfer to a critical care unit, where they can receive intravenous positive inotropic medications (such as dopamine, dobutamine, norepinephrine or milrinone) or mechanical assist devices such as an intra-aortic balloon pump or extracorporeal membraneous oxygenation
- Ensure that the patient is in bed, with the head of bed either flat or slightly elevated—the patient will probably not be able to tolerate a totally flat bed, especially if they are experiencing respiratory distress. Avoid Trendelenburg position. There is no current evidence to support its efficacy in hypotensive patients, and in fact it may be harmful
- Work collaboratively with other members of the health care team—medicine, respiratory therapy, the code team—to ensure that the patient receives timely care and is transferred to the appropriate critical care area
Health team Establish a systematic approach to report cardiac changes to appropriate #
In order to communicate effectively to the resident or the staff man, one must use a systematic approach to report cardiac condition. The SBAR is one such system. Table 4 demonstrate an example of the SBAR system.

Participate in the emergency response #
Participate in the emergency response to the resuscitation of a cardiac patient as a member of the health care team
When the patient progresses to (cardiogenic shock) a code situation, the role of the primary nurse is to activate the code button at the bedside, get other staff to bring the ward code cart to the bedside and stay with the patient to ensure ABC is maintained.

Airway (A)
- Maintain patient’s airway with oral airway
Breathing (B)
- Use ambu bag with 100% oxygen to assist ventilation if patient is not breathing or having difficulty breathing
Circulation (C)
- Check BP and HR and if no palpable pulses check carotid pulses before starting CPR.
- Ensure a running IV line exist to administer medications
- Delegate duties to other staff such as recording the code, gathering supplies and calling the admitting service/doctor
- Prepare the patient’s chart to be available to the code team.
- Provide the code team with a brief overview of the patient’s history and the event that led to the code and any interventions rendered.
- Assist the code team to get supplies and send laboratory specimens
- Prepare and assist the code team to move the patient to ICU.
- Notify the family of the event and the transfer to ICU.
Remember, do NOT leave the room when the code team arrives.
Simulation of Pre-code Management with a Cardiac Patient #
The screen below is an interactive simulation that explores the management of a cardiac pre-code event.
References #
Archar, S., Kundu, S., & Norcross, W. (2005). Diagnosis of Acute Coronary Syndrome. American Family Physician, 72(1), p. 119-126.
Bench, S. (2004). Clinical skills: Assessing and treating shock: A nursing perspective.British Journal of Nursing, 13(12), p. 715-721.
Bridges, N., & Jarquin-Valdivia, A.A. (2005). Use of the Trendelenburg position as the resuscitation position: To T or not to T? American Journal of Critical Care, 14(5), p. 364-367.
Cardiovascular Care Made Incredibly Easy (2005). Donofrio, J., Haworth, K., Schaeffer, L., & Thompson, G., Editors. Philadelphia: Lippincott Williams & Wilkins.
Dean, R. (2005). Emergency first aid for nurses. Nursing Standard, 20(6), p. 57-65.
Devon, H. & Ryan, C. (2005). Chest Pain and Associated Symptoms of Acute Coronary Syndromes, 20(4), p. 232-238
Ducas, J., & Grech, E.D. (2003). Percutaneous coronary intervention: Cardiogenic shock. British Medical Journal, 326, p.1450-1452.
Fitchett, Goodman & Langer (2001)
Hasdal, D., Topol, E.J., Califf, R.M., Berger, P.B., & Holmes, D. R. Jr. (2000). Cardiogenic shock complicating acute coronary syndromes. Lancet, 356, p. 749-756.
Holcomb, S.S. (2002). Cardiogenic shock: A success story. Dimensions of Critical Care Nursing, 21(6), p. 232-235.
Holcomb, S.S. (2002). Helping your patient conquer cardiogenic shock. Nursing2002, 32(9), p. 32cc1-32cc6.
Hollenberg, S.M., Kavinsky, C.J., & Parillo, J.E. (1999). Cardiogenic shock. Annals of Internal Medicine, 131(1), p. 47-59.
Lackey, S. (2006). Suppressing the scourge of AMI. Nursing2006, 36(5), p. 37-41.
McSeeney Et al (2003).
Meador, B. (1982). Cardiogenic shock: Help break the vicious cycle. RN, April, p. 38-42.
Revelly, J.P., Tappy, L., Martinez, A., Bollmann, M., Cayeux, M.C., Berger, M.M., & Chioléro, R.L. (2005). Lactate and glucose metabolism in severe sepsis and cardiogenic shock. Critical Care Medicine, 33(10), p. 2235-2240.
Revision: Saturday Feb 4, 2017 – 00:09.
Last Modified: 0.
Release Date: Tuesday Mar 20, 2007 – 18:00.